-

© Universität Bielefeld

Research
The primary material analyzed by genome informatics are genomic sequences. Beyond the acquisition and basic analysis of these data, the next challenge is to extract the higher-level information encoded in them, which poses the need for sound mathematical models, efficient algorithms, and user-friendly software.
Research in the Genome Informatics group spans a broad spectrum in this exciting field, from the low level of DNA sequence comparison up to the higher levels of comparative genomics, and making better infrastructures.
Efficient Comparison of DNA Sequences (aka team kmers)
Improving search engines for DNA sequences
The amount of publicly available sequencing data is growing faster than computational power. Searching for a sequence of interest among datasets is a fundamental need; however, no method scales to the dozens of petabytes of data already available today. Thus, new computational methods are required to perform a search against datasets.
Queries on large-scale datasets are usually done by indexing all k-mers (words of length k) from sequences. These k-mers are then typically indexed in an Approximate membership query (AMQ) data structure. The proportion of shared k-mers between the indexed datasets and the AMQ then gives an overview of the presence of the query in a dataset. To index a set of sequences, AMQ data structures typically require less space than the original set. However, AMQ data structures suffer from a non-null false positive rate, which biases downstream analysis.
AMQ data structures can be generalized for additionally recording the abundance of indexed elements, they are then called “counting AMQ” data structures. The abundance information is crucial for many biological applications such as transcriptomics or metagenomics. However, counting AMQs data structures suffer from false positives and overestimated calls.
We propose strategies to reduce the false positive rate and overestimation rate of both AMQ and counting AMQ data structures.
Fast Heuristic Local Alignment Search in Pangenome Graphs

The advent of High Throughput Sequencing raises a major concern about storage and indexing of data produced by these technologies. Large-scale sequencing projects generate an unprecedented volume of genomic sequences ranging from tens to several thousands of genomes per species. These collections contain highly similar and redundant sequences, also known as pangenomes. Pangenomes are oftentimes represented by graphical data structures such as colored de Bruijn graphs (CDBGs) in which vertices represent colored k-mers, words of length k associated with the genomes in which they occur. CDBGs may also be compacted by merging vertices of unique non-branching paths. Representing pangenomes as compacted CDBGs is beneficial, but it requires modifications of methods to query the data. We study the problem of finding high scoring local alignments between a query sequence and a compacted CDBG that are likely to represent sequence homology. Our work is in line with the popular BLAST algorithm. Our method is published in Bioinformatics. An implementation and further documentation are available here.
Sequence-Based Pangenomic Core Detection
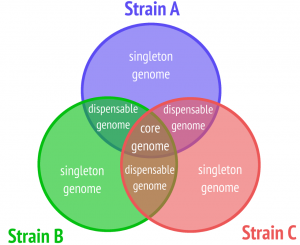 One of the most basic kinds of analysis to be performed on a pan genome is the detection of its core, i.e., the information shared among all members. Pangenomic core detection is classically done on the gene level and many tools focus exclusively on core detection in prokaryotes.
We are working on a new method for sequence-based pangenomic core detection. Our model generalizes from a strict core definition allowing us to flexibly determine suitable core properties depending on the research question and the data set under consideration. We propose an algorithm based on a CDBG that runs in linear time with respect to the number of k-mers in the graph. An implementation of our method is called Corer. Due to the usage of a CDBG, it works alignment-free, is provided with a small memory footprint, and accepts as input assembled genomes as well as sequencing reads. Corer has been presented as RECOMB-seq in 2022 and a manuscript is published in iScience.
One of the most basic kinds of analysis to be performed on a pan genome is the detection of its core, i.e., the information shared among all members. Pangenomic core detection is classically done on the gene level and many tools focus exclusively on core detection in prokaryotes.
We are working on a new method for sequence-based pangenomic core detection. Our model generalizes from a strict core definition allowing us to flexibly determine suitable core properties depending on the research question and the data set under consideration. We propose an algorithm based on a CDBG that runs in linear time with respect to the number of k-mers in the graph. An implementation of our method is called Corer. Due to the usage of a CDBG, it works alignment-free, is provided with a small memory footprint, and accepts as input assembled genomes as well as sequencing reads. Corer has been presented as RECOMB-seq in 2022 and a manuscript is published in iScience.
Pangenome Openness

The field of pangenomics emerged from the study of bacterial genomes, initially defining the pangenome as the comprehensive set of genes within a species. Early research discovered substantial differences in pangenome sizes across species, introducing the concept of pangenome openness. This concept reflects a species' capacity for colonizing new environments and its genetic diversity. Given the rapid increase in genomic data and the time-consuming steps required by traditional gene-based methods – such as annotation, gene homology identification, and the need for fully assembled genomes – we propesd an alternative to estimate pangenome openness with the use of k-mers, short DNA sequences of fixed length. We developed a tool called Pangrowth, which efficiently estimates this openness.
Computational Comparative Genomics
Comparative Pangenomics

Genome rearrangements have been studied extensively in theoretical works of Comparative Genomics. These results however, have only been applied on a limited scale to real genomes. The continuing progress of sequencing projects and technology made more and more high quality genomes available and enabled even Pangenomic analyses, that is, analyses that include all availailable genomes of a species. Pangenomics and theoretical Rearrangement Studies utilize remarkably similar graph data structures. Given the abundance of theoretical results in Comparative Genomics, it is likely that many of these results can be applied in Pangenomics. Conversely, the abundance of practical results in the construction of Pangenome graphs can likely contribute to these theoretical results seeing more real world applications.
Orthology Inference via Large-scale Rearrangements

Computing distances based on large-scale rearrangements between two family-annotated genomes (Bohnenkämper et al., 2021) was converted into a method that takes genome rearrangements into consideration for inferring gene orthologies of two genomes (Rubert, Martinez & Braga, 2021). For a set of k genomes, the inference of pairwise gene orthologies is the core of OrthoFFGC, a tool for inferring gene families across the k input genomes (Rubert, Doerr & Braga, 2021; Rubert & Braga, 2023; OrthoFFGC-website).
Sequence-based Phylogenomics
 Comparative genomics often involves the reconstruction of phylogenies. The ever-increasing number of available genomes, many of which are published in an unfinished state or lack sufficient annotation, poses challenges to traditional phylogenetic inference methods that rely on the comparison of marker sequences.
Whole-genome approaches have emerged as a solution to these challenges, but as these approaches are based on pairwise comparisons between genomes, their runtime increases quadratically with the number of input sequences, making them unsuitable in large-scale scenarios.
Comparative genomics often involves the reconstruction of phylogenies. The ever-increasing number of available genomes, many of which are published in an unfinished state or lack sufficient annotation, poses challenges to traditional phylogenetic inference methods that rely on the comparison of marker sequences.
Whole-genome approaches have emerged as a solution to these challenges, but as these approaches are based on pairwise comparisons between genomes, their runtime increases quadratically with the number of input sequences, making them unsuitable in large-scale scenarios.
SANS (tool-website; Rempel and Wittler, 2021; Wittler, 2020) is a whole-genome based, alignment- and reference-free approach that does not rely on a pairwise comparison of genomes. In a pangenomic approach, evolutionary relationships are determined based on the similarity of the whole sequences. Sequence segments (k-mers) shared by a subset of genomes are interpreted as a phylogenetic split indicating the closeness of these genomes and their separation from the other genomes.
Evolution of Gene Clusters
 We integrate the concept of conserved gene clusters into the framework of phylogenetics. Here, the focus is not any more on the discovery of new gene clusters, but on their evolution. Given the topology of a phylogenetic tree and the gene orders of the leaf nodes, our methods reconstruct ancestral gene orders at the internal nodes under different evolutionary (rearrangement) models (see Rococo, RINGO, PhySca).
We integrate the concept of conserved gene clusters into the framework of phylogenetics. Here, the focus is not any more on the discovery of new gene clusters, but on their evolution. Given the topology of a phylogenetic tree and the gene orders of the leaf nodes, our methods reconstruct ancestral gene orders at the internal nodes under different evolutionary (rearrangement) models (see Rococo, RINGO, PhySca).
In addition, the development of ancient DNA (aDNA) sequencing led us to the problem of integrating this additional data in the reconstruction of ancestral genomes, aiming to scaffold fragmented aDNA assemblies and to improve the global reconstruction of all ancestors in the phylogeny.
Infrastructure
SANS-ambages
 We maintain a software tool for alignment-free, whole-genome based phylogenomics implementing the SANS approach (see above: “Sequence-based Phylogenomics”). The current version “SANS ambages” (abundance-filter, multi-threading and bootstrapping
on amino-acid or genomic sequences) provides several new features:
besides processing DNA sequences (whole genomes or assemblies), SANS ambages can also work on amino acid level taking protein
sequences (translated or untranslated) as input. Further, the ability to process read data has been enhanced by the option to filter out low-abundant sequence segments. Multiple input sequences can be processed in parallel, and bootstrapping allows to augment the output with confidence values.
SANS is hosted as a de.NBI service and can easily be obtained from our Gitlab repository.
We maintain a software tool for alignment-free, whole-genome based phylogenomics implementing the SANS approach (see above: “Sequence-based Phylogenomics”). The current version “SANS ambages” (abundance-filter, multi-threading and bootstrapping
on amino-acid or genomic sequences) provides several new features:
besides processing DNA sequences (whole genomes or assemblies), SANS ambages can also work on amino acid level taking protein
sequences (translated or untranslated) as input. Further, the ability to process read data has been enhanced by the option to filter out low-abundant sequence segments. Multiple input sequences can be processed in parallel, and bootstrapping allows to augment the output with confidence values.
SANS is hosted as a de.NBI service and can easily be obtained from our Gitlab repository.
SOCKS & PanBench
A major challenge in computational pangenomics is the memory- and time-efficient analysis of multiple genomes in parallel. Many software tools used in the current research follow the idea of colored k-mer sets to efficiently index and query a large collection of strings, but use different strategies when it comes to the implementation. The aim of this project is to evaluate the different solutions and establish a standard interface.
The SOCKS interface (Software for colored k-mer sets) defines a common set of core features and standard input and output formats that software tools in computational pangenomics should implement. It aims to enhance the comparability and interoperability of these tools for the benefit of both developers and users. A detailed description of the interface including some examples can be found on the dedicated project page.
PanBench (Pangenomics Benchmark and Workbench) is an open catalog of software tools for computational pangenomics and is an example of what such a common interface makes possible. It allows users and developers to search for tools by different criteria, compare the performance of these tools, and test each tool in a user-friendly web interface before downloading and installing the software on their local machine.
Panacus
Pangenomics aims to capture the collective genomic diversity of taxonomically related genomes, typically from the same species, offering potential insights for medicine and biology. Initially defined as collections of genes, pangenomes are now more accurately represented through variation graphs. These graphs illustrate genomes by using nodes to depict shared sequences, edges to denote sequential connections, and paths to reconstruct the original genomes. Despite their benefits, there is a lack of scalable software for essential analyses, such as estimating the core genome size and evaluating the extent of genomic variability (pangenome growth). To address this need, we developed Panacus (pangenome-abacus), a tool designed for efficient data extraction from GFA files, the standard for pangenome graphs. Panacus facilitates quick generation of pangenome growth and core curves, handling millions of nodes in variation graphs within minutes.

MicroGenUniBi and NFDIs
Under the acronym “MicroGenUniBi” (de.NBI-Resource Center for Microbial Genome Research in Biotechnology and Medicine at Bielefeld University), we provide services as partner of the de.NBI network. MicroGenUniBi ensures the operation of the de.NBI cloud site at Bielefeld University which involves the installation and maintenance of hardware components, the configuration of software and the (re)certification of the cloud infrastructure. Furthermore, MicroGenUniBi provides services in microbial bioinformatics and offers training courses introducing to the usage of these services on a regular basis. Currently, provided services cover areas of pangenomics, metagenomics and multiomics analyses. In particular, the area of pangenomics is represented by the services PLAST for pangenome alignment, Corer for the prediction of a pangenome’s core genome and SANS for phylogenetic analyses (see also above). The programs QuPE, MeltDB and Fusion cover the area of multiomics analyses. In addition, MicroGenUniBi provides various software solutions, e.g. for the computation of alignments, the comparison of genomes and RNA structure prediction which are collectively available from the platform BiBiServ. Moreover, the web-based software BIIGLE for collaborative image and video annotation is offered, specifically for research in biodiversity. We are also involved in the National Research Data Infrastructure Germany (NFDI) initiative. In particular, we contribute to NFDI4Biodiversity and NFDI4Microbiota.
![]()


 Except where otherwise noted, content on this wiki is licensed under the following license: CC Attribution-Share Alike 4.0 International
Except where otherwise noted, content on this wiki is licensed under the following license: CC Attribution-Share Alike 4.0 International